
How to Get an Ehlers-Danlos Syndrome Diagnosis
Editor's Note
The Mighty’s Condition Guides combine the expertise of both the medical and patient community to help you and your loved ones on your health journeys. For the Ehlers-Danlos syndrome (EDS) guide, we interviewed three medical experts, read numerous studies and surveyed more than 750 people living with EDS. The guides are living documents and will be updated with new information as it becomes available.
{kind=link}
How Do You Get an Ehlers-Danlos Syndrome Diagnosis?
To get an Ehlers-Danlos syndrome (EDS) diagnosis, your doctor will review your medical and family history, perform a physical examination and in some cases will order genetic testing. Of the 13 EDS subtypes, 12 can be confirmed with genetic testing. However, doctors don’t know which genes cause the most common type of EDS, hypermobile Ehlers-Danlos syndrome (hEDS). To diagnose hEDS, doctors rely on your history and a physical examination.
• What is Ehlers-Danlos Syndrome?
• What Are Common Ehlers-Danlos Syndrome Symptoms?
Learn More About Ehlers-Danlos Syndrome: Overview | Symptoms | Treatment | Resources

Medically reviewed by Shanda Dorff, M.D., FAAFP
What to Expect When You Suspect EDS | Hypermobile Ehlers-Danlos Syndrome | Genetic Diagnosis For Other Subtypes | Other Conditions to Consider
Understanding EDS: Getting an Ehlers-Danlos Syndrome Diagnosis
Though the symptoms of Ehlers-Danlos syndrome (EDS) were first observed as far back as the 1600s and named in the 1900s, it’s a condition that flew under the radar for many years. As their understanding of Ehlers-Danlos syndrome (EDS) has increased, doctors and scientists realized they needed a better system to group the symptoms of each EDS subtype.
In 1967, the first three types of EDS were classified by A.P. Barabas followed by Beighton in 1969, who outlined five types of the condition. Around 1988, doctors used “Berlin Nosology” to guide diagnosis, which included 11 EDS subtypes.10 This document was updated in 1998 and became the “Villefranche Nosology.” Doctors slimmed the number of EDS subtypes down to six major types.
However, the old 1998 criteria didn’t take into account some of the rarer but clinically distinct subtypes we now recognize. In 2017, doctors and scientists came together to create the 2017 International Classification for the Ehlers-Danlos Syndromes.10 The new criteria established 13 EDS subtypes and outlines the clearest picture of diagnosis for doctors. It outlines the physical signs (observable evidence of the condition) and symptoms (what you experience) associated with each type of EDS and the genes identified to cause each of these types.
When your doctor reviews your symptoms in relation to the new international EDS criteria, they record your major and minor symptoms. Each subtype of EDS has major criteria, meaning symptoms that more than 80% of people with that subtype will have.10 The minor criteria include less consistent signs and symptoms that will help your doctor make an accurate diagnosis as well as guide treatment. In total, there are over 100 separate signs and symptoms across all 13 subtypes of EDS.
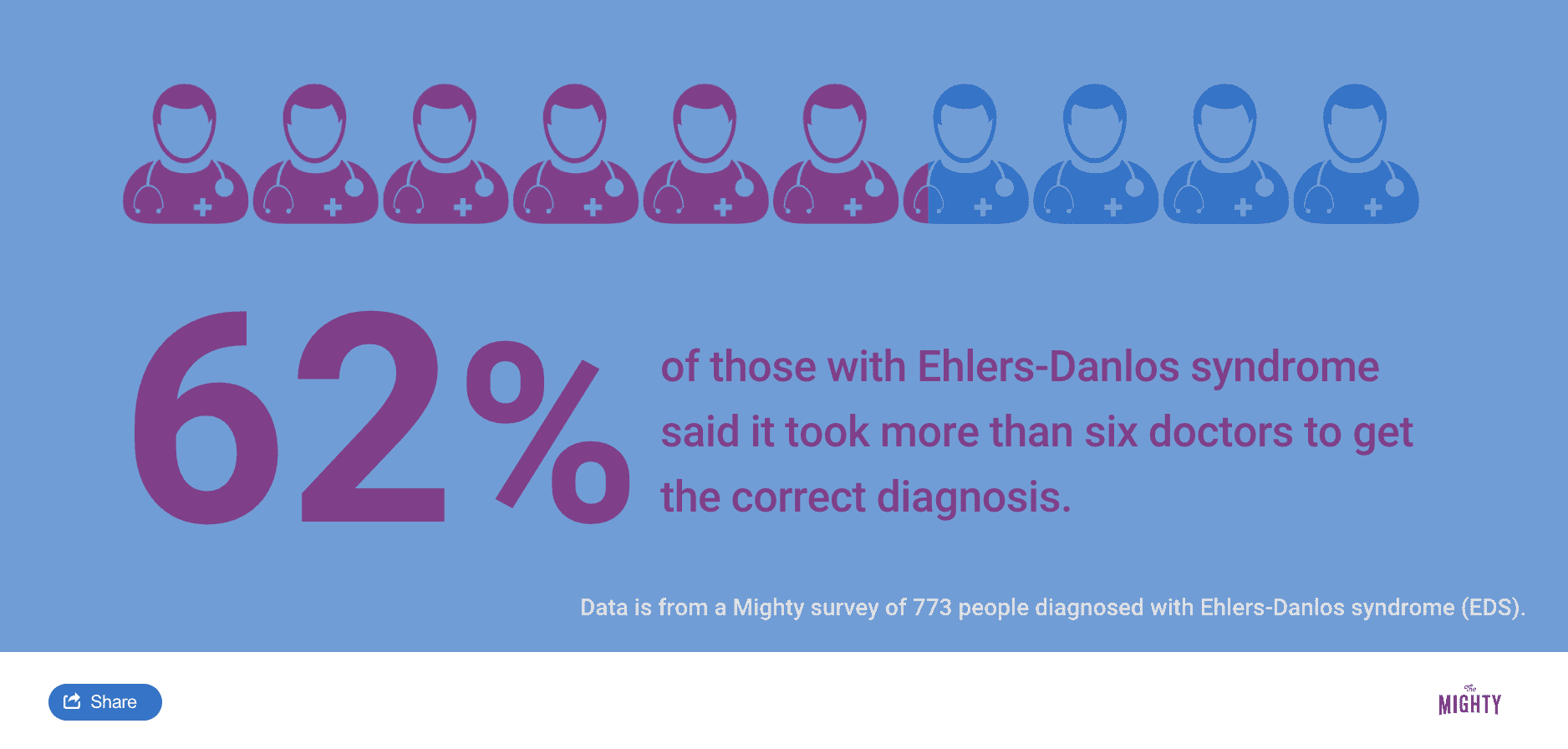
What to Expect When You Suspect EDS
EDS can be a tricky condition to diagnose because of the wide range of symptoms. EDS was also considered “rare” for a long time. Most subtypes of EDS are indeed quite rare — sometimes only one case out of every 1 million people. However, experts now realize EDS is much more common than they thought. It’s estimated that as many as 1 out of every 3,000 people have hypermobile Ehlers-Danlos syndrome, the most common subtype.1
When your doctor suspects EDS, there are two ways they might make a diagnosis. For 12 of the 13 subtypes of EDS, you may be referred to a geneticist or your doctor may order a genetic test to determine which of your collagen or connective tissue genes might show a change. Depending on the genes affected, your doctor will be able to confirm which subtype of EDS you have as well as potentially rule out other similar conditions. However, scientists haven’t figured out which gene or genes cause hypermobile Ehlers-Danlos syndrome (hEDS), so your doctor will make a clinical diagnosis based on your signs and symptoms. In some cases, your physical therapist might be the first professional to suspect you have EDS because they recognize joint hypermobility during assessment.

This information is still quite new, so many doctors haven’t caught up on what EDS looks like. For this reason, many people wait years until they’re properly diagnosed. They may be misdiagnosed completely more than once, or worse, their symptoms dismissed. It may also take some time to manage other complications or get to the bottom of the underlying complications that weren’t evaluated at a younger age.
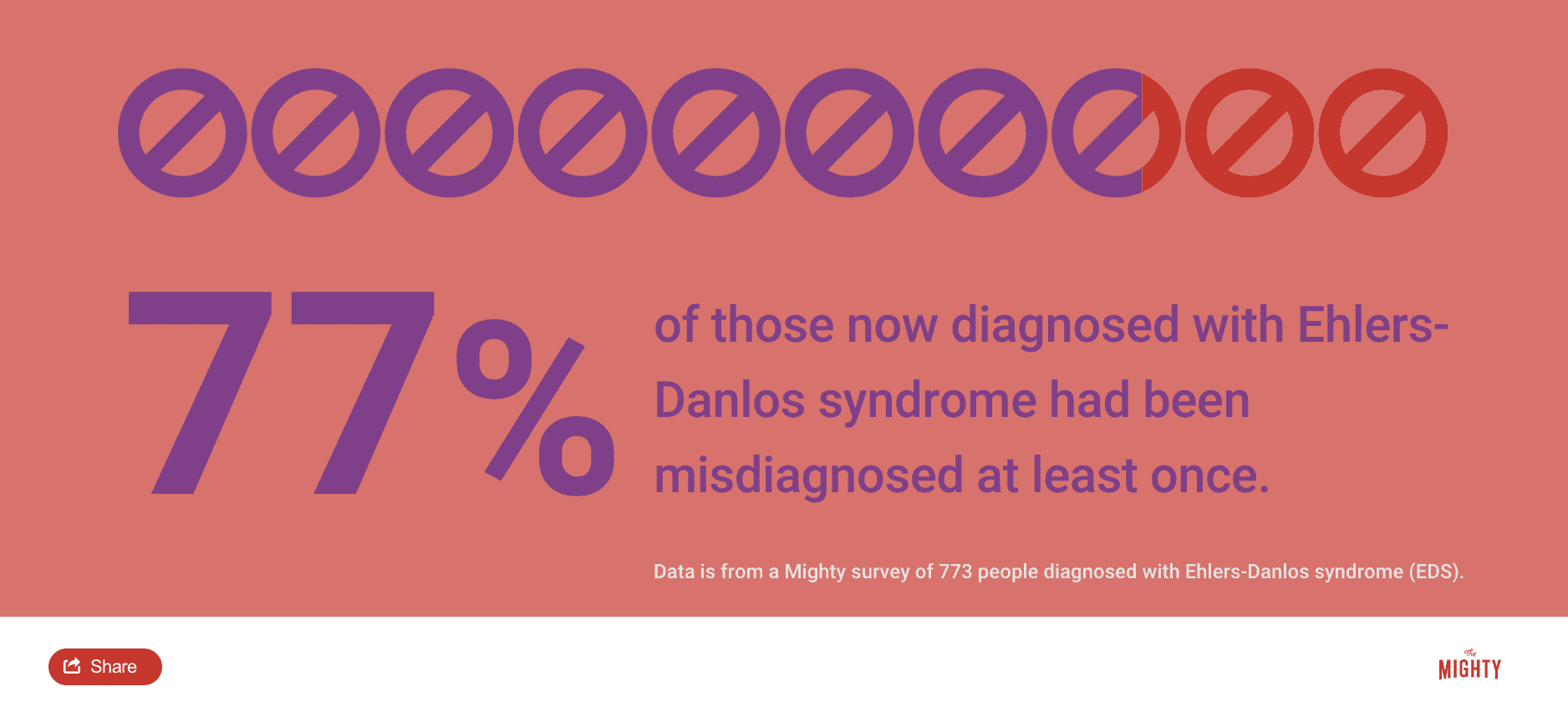
Hypermobile Ehlers-Danlos Syndrome
Hypermobile Ehlers-Danlos syndrome (hEDS) is the only subtype of EDS for which there isn’t a genetic test yet. Your doctor must review your medical history, complete a physical examination and compare your signs and symptoms to the official 2017 International Classification for the Ehlers-Danlos Syndromes diagnostic criteria. In order to make an hEDS diagnosis, your doctor will make sure you meet all three of the criteria groups.10
Criteria 1: Generalized Joint Hypermobility
The first criteria a doctor will check is generalized joint hypermobility. This means many of your joints move beyond what’s considered average. If you only have one overly flexible joint, that’s not enough to meet the criteria for an hEDS diagnosis. Your doctor will assess your joint hypermobility by using the Beighton Score system, which measures your flexibility in different joints in your hands, arms, legs and lower spine.
Beighton Scoring System
The Beighton Score is a non-invasive test during which your doctor will ask you to perform a series of five different movements to measure your flexibility.6 This includes your pinky fingers, thumbs, elbows, knees and your lower spine. For some parts of the test, your doctor may need to measure the precise angle of your joints with a tool called a goniometer, which is like a protractor for your joints.
For example, in one test to determine your Beighton score, you’ll sit with your arm sticking straight out in front of you so it forms a 90-degree angle with your body. Turn your hand so the palm faces the floor. Now gently pull your thumb toward your forearm, which is facing the floor. If you are able to pull your thumb back to make it touch your arm, you get one point for the positive result.
You will continue this evaluation through four movements on both the left and right sides of your body in addition to your spine to determine your final score. Each time you test “positive,” you assign a point, with a max number of nine points.
The Beighton score is a bit inconsistent from person to person based on your age, gender, height and weight, so it’s only one piece of your total diagnosis. However, for young people who have not gone through puberty, a score of six or more is generally considered a positive diagnosis of generalized joint hypermobility (GJH). For adults past puberty up until age 50, GJH is confirmed with a score of five or more, and after age 50, four or more points are required.
Five-Point Questionnaire
In some cases, the Beighton scoring system isn’t helpful. For example, if you had joint surgery or use a wheelchair, you may not be able to complete the movements of the Beighton Score, or the test is not accurate. Your doctor can also use a five-point questionnaire to assess the degree of your joint flexibility that takes into account what you can do now and what you could do in the past. The five-point questionnaire is not recommended for children. The five questions you will be asked are:
- Can you now (or could you ever) place your hands flat on the floor without bending your knees?
- Can you now (or could you ever) bend your thumb to touch your forearm?
- As a child, did you amuse your friends by contorting your body into strange shapes or could you do the splits?
- As a child or teenager, did your shoulder or kneecap dislocate on more than one occasion?
- Do you consider yourself “double-jointed”?
If you answer “yes” to two or more questions, that’s enough to qualify as having hypermobile joints. Because the Beighton score is different person to person, the five-point questionnaire may also be used in combination with your Beighton score when the Beighton score is one point less than the threshold used for diagnosis (a Beighton score of four for adults, five for children or three for adults over the age of 50). In this case, if you answered “yes” to two questionnaire questions, your doctor would still consider that confirmation of hypermobile joints.
Criteria 2: Other Symptoms
Criteria 2 for an hEDS diagnosis looks at both your other signs and symptoms and your family’s medical history, including other relatives who have been diagnosed with hEDS. There are three parts to criteria 2 — parts A, B and C. When considering an hEDS diagnosis, you need to meet the criteria for at least two of the three parts.
In part A, your doctor will note your other symptoms from a list of 12 signs associated with hEDS. To “test positive” for hEDS in part A, your doctor will be looking for you to meet at least five of the following signs or symptoms:
- Skin that feels soft or velvety
- Some extra stretchiness in your skin
- Stretch marks on your body not caused by weight gain or loss
- Painful bumps on your heels called piezogenic papules caused by fat that has pushed through internal layers of your skin
- More than one groin or abdomen hernia. A hernia happens when organ tissue or fat pushes through tissue inside your body where it shouldn’t and can cause a burning or sharp pain in the area
- “Dented” or “pitted” scarring, called atrophic scarring, in at least two areas
- A pelvic floor, rectal or uterus prolapse. A prolapse occurs when the connective tissue holding an organ in places disconnects and the organ — a part of your colon or uterus — comes outside of your body
- Dental crowding or your teeth don’t seem to have enough room in your mouth
- Disproportionately long fingers in comparison to the length of your palm, sometimes called “spider fingers” (more often associated with the connective tissue disorder Marfan syndrome than hEDS)
- Your arm span — the distance from fingertip to fingertip when you hold your arms straight out — is more than 1.05 times your height (more common with Marfan syndrome than hEDS)
- Mitral valve prolapse, which can be described as when the doors between the chambers of your heart do not open and close appropriately and are a little loose so they extend or dangle a bit beyond their tracts and may cause some problems from not working right, like an irregular heartbeat, chest pain, shortness of breath or dizziness
- An enlarged or dilated aorta (the main artery in your body) that doctors would measure as being at least two standard deviations above average
Part B asks about your family’s health history. If at least one of your first-degree relatives — your parents, siblings or children — have a diagnosis or meet these criteria for hEDS, that will factor into an hEDS diagnosis.
Finally, your doctor will ask about “musculoskeletal complications” in part C. Musculoskeletal refers to your bones, muscles and joints. In Part C, there are three complications that can be considered for diagnosis, and for hEDS, you must meet at least one.
First, you may have musculoskeletal pain in your bones, muscles or joints in at least two of your limbs, like your arms and legs. Second, you could have chronic widespread pain that isn’t easy to pinpoint. In either case, to make a diagnosis of hEDS, the pain must occur every day for three months of longer. If you meet either of these pain categories, that’s a “positive” result for the part C criteria.
A doctor would also consider joint dislocations or joint instability a musculoskeletal complication. To count toward an hEDS diagnosis, your joint dislocations must have occurred at least three times in the same joint or two or more dislocations in two different joints at two different times. In either case, these dislocations can’t be caused by a trauma or obvious injury, like a bicycle accident, for example. Alternatively, joint complications may be general joint instability even if you don’t typically have joint dislocations, which would need to be medically confirmed in at least two separate joints.
Criteria 3: Excluding Alternate Diagnoses
The part 3 criteria for hEDS makes sure your doctor has done their due diligence when making a diagnosis. Called prerequisites, your doctor will consider three additional factors before making an hEDS diagnosis. First, to be categorized as hEDS, your doctor will look for other signs and symptoms typical of other subtypes of EDS, like fragile skin, severe prolapse, a perforation or unexpected vascular signs. If your doctor identifies any of these signs or symptoms, they will want to rule out the other subtypes of EDS.
Secondly, your doctor should rule out other common genetic and connective tissue disorders. This could include autoimmune disorders like lupus or rheumatoid arthritis, and conditions that fall under the banner of rheumatology like fibromyalgia, HIV or Lyme disease. While doctors can test for most of these conditions, it’s still tricky because there is often overlap between symptoms. Plus, you can have EDS and another condition. If this is the case, to diagnose hEDS, you will need to meet all the official diagnostic criteria.
Finally, your doctor should rule out alternative diagnoses that include joint hypermobility because of muscle weakness or a connective tissue issue. This could mean other subtypes of EDS along with disorders like Bethlem myopathy, Loeys-Dietz syndrome, Marfan syndrome or skeletal dysplasias that affect bone growth. Excluding these other conditions can be difficult because there are not tests for all of them. Doctors will have to carefully consider your signs and symptoms, medical history, genetic testing and other available lab tests.

Genetic Testing
There is no genetic test to confirm hEDS because researchers don’t know which gene causes the collagen protein differences in hEDS. Geneticists theorize it could be more than one gene or several different genes with mutations or changes that interact in a way to cause hEDS.1 Another theory suggests it might be caused not by your genes or DNA itself, but something happens as the information is processed by your biology. Genetic testing can be useful to exclude the 12 other subtypes of EDS just in case.1
Though science has evolved significantly in recent years, there are still limits to how much information we can discover with current DNA technologies. As science advances and new technologies are developed, scientists are hopeful they will discover the genetic cause of hEDS, so it’s easier to diagnose through genetic testing.
Hypermobile EDS Versus Hypermobility Spectrum Disorders
Sometimes your doctor may suggest another hypermobile condition generally called hypermobility spectrum disorders (HSD). The label was created as an alternative diagnosis when you don’t fulfill all the diagnostic requirements for hEDS.14 Hypermobility can be thought of on a spectrum in terms of how severe your symptoms might be. At one end of the scale, you may have flexible joints without pain or other symptoms. At the other end of the HSD spectrum, you meet the full criteria to be diagnosed with hEDS.7
The additional consideration of HSD allows doctors a kind of in-between diagnosis.14 Since doctors can’t confirm hEDS via genetic testing, they may opt to give you an HSD diagnosis if they’ve ruled out other conditions with joint flexibility but are unable to make a full hEDS diagnosis based on the strict criteria.
Genetic Diagnosis for Other Subtypes
Once your doctor suspects an EDS subtype based on the symptoms you report, your doctor may order a genetic test or refer you to a geneticist. Ideally, genetic testing can confirm or rule out an EDS diagnosis by looking for changes in collagen-related genes connected to the EDS subtypes. Outside of hEDS, the other 12 EDS subtypes are associated with specific, distinct genes. However, if you’re not able to get genetic testing because it’s too expensive or you can’t find a lab near where you live, your doctor can still make an EDS diagnosis based on your signs and symptoms. Having a diagnosis, with or without testing, will support your treatment, preventative measures and even your prognosis.2
When you’re sent for testing, a clinical geneticist has the expertise to determine the genes that point to an EDS diagnosis versus any of more than 200 other inherited connective tissue-related diagnoses.5 The test is non-invasive and is done by testing a blood, hair, tissue or saliva sample sent to a genetics lab.19 Geneticists can also test the amniotic fluid around a fetus during pregnancy to look at genetic material. They will usually use molecular testing, which looks at a small era of your DNA to zero in on specific genes to look for variations.9 Once the results have been finalized in the lab, the results are sent to both you and your doctor for review.
If your doctor recommends genetic testing, it can be expensive. Health insurance will sometimes cover at least some of the cost of a genetic work-up if it’s recommended by your doctor.15 In the U.S., the cost can still range from $100 to more than $2,000 or more, depending on what you need, where you go, what your insurance may cover or if you pay out of pocket.15 Know that sometimes it may be 10 times more expensive when a genetic test is billed to your insurance company because of additional administrative fees. Though if your insurance covers genetic testing, your out-of-pocket cost will usually be less than the amount billed to your insurance company.
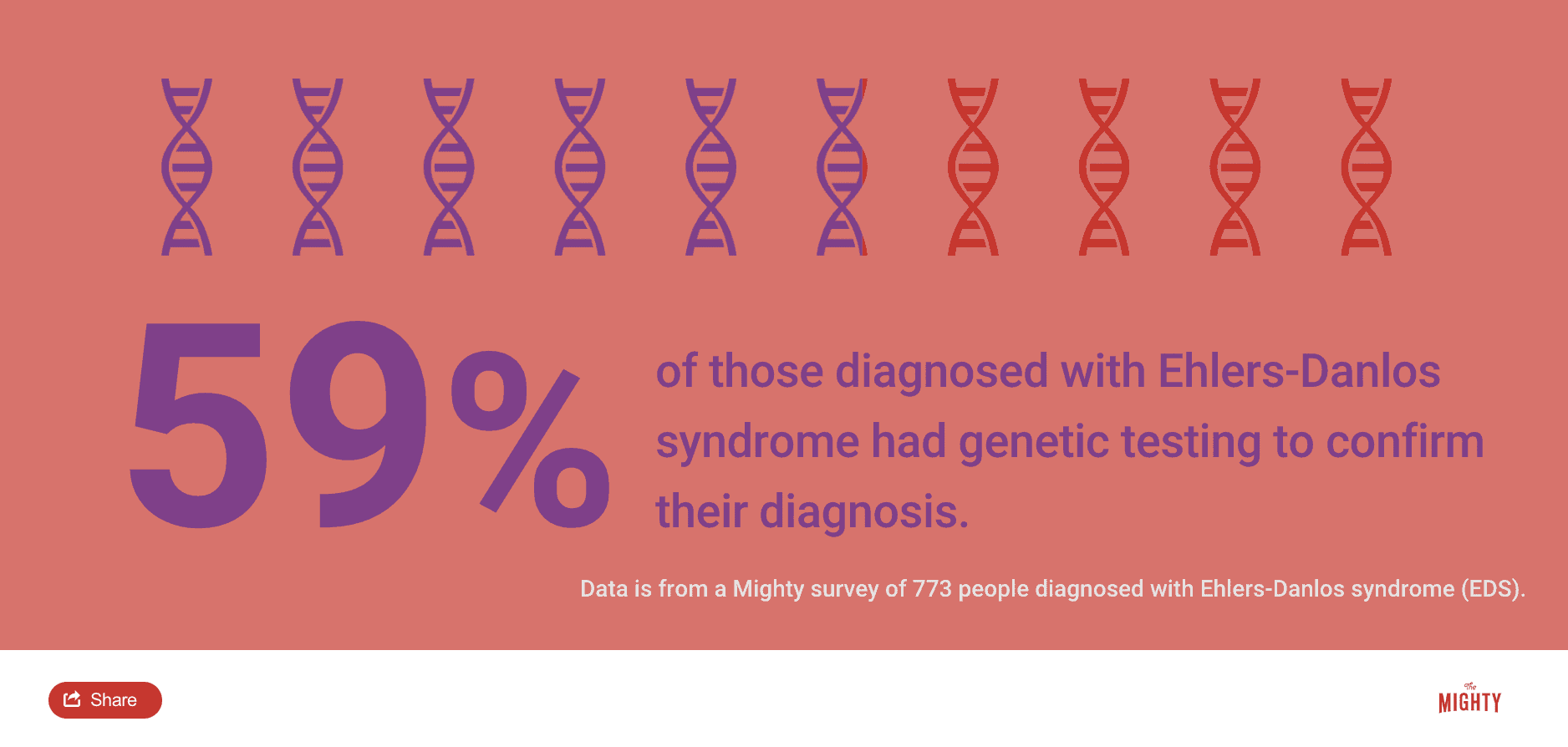
Classical Ehlers-Danlos Syndrome (cEDS)
To diagnose classical Ehlers-Danlos syndrome (EDS), a geneticist looks for a mutation in your type V collagen genes, COL5A1 and COL5A2, the root of 90 percent of cEDS cases.10 The differences in your skin, which includes stretchiness, dented or atrophic scars, easy bruising and fragile skin, are a hallmark of cEDS thanks to these genes’ involvement.
In rare instances, changes to your type I collagen genes — COL1A1 c.934C>T, p.(Arg312Cys) — may be the underlying cause of cEDS.10 If you have this genetic change, you’re more likely to have dangerous blood vessel or heart complications, so it’s important to distinguish this version of cEDS right down to the genetic level.
Vascular Ehlers-Danlos Syndrome (vEDS)
Because vascular Ehlers-Danlos syndrome (vEDS) impacts your blood vessels and is associated with ruptured arteries or organ failures, it’s especially important to diagnose as soon as possible. The gene most often linked to vEDS is COL3A1, a gene that encodes your type III collagen.10 In rare cases, a geneticist might find changes in your type I collagen genes, COL1A1, which is linked to more vascular system issues and a more severe course of illness.10 This type of EDS is also associated with multiple family members during at young ages from complications such as aneurysms or ruptures.
Classical-like EDS (clEDS)
To test for classical-like Ehlers-Danlos syndrome (clEDS), geneticists will look at your TNX-related genetic material. TNX is a type of protein linked to your muscle tissue, tendons, ligaments and skin. Mutations in your TNXB gene cause TNX to be absent completely, which leads to clEDS.3 This gene is autosomal recessive, which means the gene may skip a generation because you’d need to a copy of the changed gene from both of your parents to get clEDS, as opposed to only one parent in many of the other types.
Cardiac-valvular EDS (cvEDS)
Cardiac-valvular Ehlers-Danlos syndrome (cvEDS) has been linked to not having proα2‐chain of type I collagen because of changes in your COL1A2 gene. So far doctors have identified seven different mutations that can cause cvEDS, but they only occur in your COL1A2 gene. This subtype of EDS is very rare, with only six patients from five families confirmed to have the condition.3 Because this EDS type is autosomal recessive the condition can skip a generation since you need to get a changed version of the gene from both of your parents.
Arthrochalasia EDS (aEDS)
When testing for arthrochalasia Ehlers-Danlos syndrome (aEDS), your geneticist will look for changes in your COL1A1 and COL1A2 genes that cause all or part of exon 6 to be deleted. So far less than 50 patients have a confirmed aEDS diagnosis.3
Dermatosparaxis EDS (dEDS)
Dermatosparaxis Ehlers-Danlos syndrome (dEDS) was first identified in the early 1970s because animals get the condition as well.3 It’s caused by a change in the ADAMTS2 gene, the gene behind the procollagen I N‐proteinase that’s essential for healthy collagen proteins and therefore connective tissue. Notably, of the less than 20 patients known to have this rare EDS subtype, all diagnoses were made between birth and 13 years old.3 Dermatosparaxis EDS is also autosomal recessive so you’d need the changed ADAMTS2 gene from each of your parents or you won’t get the condition.
Kyphoscoliotic EDS (kEDS)
Two genes have been linked to kyphoscoliotic Ehlers-Danlos (kEDS).10 In most patients, changes in the PLOD1 gene, which affects a procollagen enzyme crucial to the stability of your collagen, cause kEDS. Another gene, FKBP14, responsible for encoding FKBP22, has more recently been linked to kEDS. You geneticist will diagnose based on what they find in either of those genes. Though it’s linked with two different genes, kEDS is autosomal recessive, so even if you parents have it, it can skip a generation because you would have to inherit the changed gene from both of your parents as opposed to just one.
Brittle Cornea Syndrome (BCS)
Brittle cornea syndrome (BCS) may be linked to a few genes, including ZNF469, which scientists don’t know much about, or PRDM5. Geneticists have found at least one family diagnosed with BCS that doesn’t have either of these genes.10 This means another gene may also cause BCS, and there’s a chance you may be diagnosed with BCS even without genetic confirmation. If only one of your parents has this type of EDS, there’s a chance you may not get the condition because it is autosomal recessive and requires each of your parents to pass along a changed collagen gene.
Spondylodysplastic EDS (spEDS)
To diagnose spondylodysplastic Ehlers-Danlos syndrome (spEDS), your geneticist will investigate three different genes that affect your connective tissue: B4GALT7, B3GALT6 or SLC39A13.10 Your doctor may find clues about which of the three genes are implicated based on slightly different spEDS minor symptoms associated with each gene. It’s also autosomal recessive, so you will only get spEDS if both of your parents also have one of the associated changed genes.
Musculocontractural EDS (mcEDS)
Musculocontractural Ehlers-Danlos syndrome (mcEDS) has been associated with a few different genes, including several mutations in the CHST14 gene, DSE gene differences and changes in the D4ST1 gene. These changes are autosomal recessive, which means in order for you to get this type of EDS, both of your parents would have to carry the gene as well, otherwise, it could skip a generation. In the past, three separate conditions were outlined based on the same deficiency related to the D4ST1 gene, so you’ll want to be sure your doctor is referencing the most current 2017 information about mcEDS.3
Myopathic EDS (mEDS)
Geneticists can identify if you have myopathic Ehlers-Danlos syndrome (mEDS) by testing for changes in your COL12A1 gene, associated with type XII collagen. This type of EDS could be either autosomal dominant or recessive, which means you could get it if just one of your parents has the changed genes or both of your parents must each pass on the gene. Therefore, mEDS can “skip” a generation depending on what pattern of inheritance you have. Your doctor will definitely want to test your genes because mEDS can look similar to other disorders. Bethlem Myopathy and Ullrich Congenital Muscular Dystrophy may have similar symptoms, but these disorders are linked to type XI collagen, not type XII like mEDS.10
Periodontal EDS (pEDS)
Periodontal Ehlers-Danlos syndrome (pEDS) is rare, so to make a diagnosis a geneticist will need to test for variations in your C1R or C1S genes, which can indicate you have the condition.3
Other Conditions to Consider
When your doctor considers a diagnosis of EDS, they will rule out other conditions with overlapping symptoms. They should consider autoimmune, dysautonomia and other musculoskeletal conditions carefully.2 And don’t forget, you can have more than one condition at the same time, including EDS. The following conditions are ones that doctors will commonly rule out, may misdiagnose or look for in combination with an EDS diagnosis.
Autoimmune Diseases
Autoimmune diseases occur when your immune system can’t tell the difference between healthy and unhealthy cells, so it becomes overly aggressive or progressive to the point your immune system also attacks your healthy cells without realizing it’s done so. Before it was well understood, EDS was confused as an autoimmune disease such as rheumatoid arthritis or lupus because their symptoms can have so much overlap, including joint pain. EDS, however, is caused by changes in your collagen genes, which isn’t typical in autoimmune conditions. An autoimmune diagnosis can usually be identified or ruled out using blood or other lab tests.
Other Connective Tissue Disorders
EDS isn’t the only connective tissue disorder that’s inherited, and many features overlap.4 For example, Marfan syndrome can include longer than average limbs, flexible joints, flat feet and eye-related issues. Unlike EDS, however, Marfan syndrome has been linked to a different protein in your connective tissue called fibrillin-1.11 Stickler syndrome, which can cause eye problems, very flexible joints and early onset arthritis is also similar to EDS but affects different genes — COL2A1, COL11A1, COL11A2, COL9A1, COL9A2 or COL9A3.8 Genetic testing can help distinguish between these genetic connective tissue disorders.
Osteopenia/Osteoporosis
Both osteopenia (lower bone density than average for your age and sex) and osteoporosis (significant bone loss that increases your risk of fractures) can be symptoms of some EDS subtypes, but they are also independent conditions.14 These bone loss conditions can be diagnosed with a bone density scan. If they’re one of a number of your EDS-related symptoms, a geneticist or EDS-knowledgeable clinician can confirm whether or not they’re a sign of your EDS in most cases.
Dysautonomic Conditions
It’s common when you have EDS, especially the hypermobile subtype, to also live with dysautonomic health conditions.2 This group of complications are related to your autonomic nervous system, which regulates functions you’re not consciously aware of, like blood flow, breathing and your digestive system, to keep everything working as it should without you having to remember every breath or tell food which direction to go in your system. EDS patients often develop irritable bowel syndrome (IBS) or postural orthostatic tachycardia syndrome (POTS), where you feel light-headed, have head rushes when standing up or faint, with EDS. Your doctor may even consider these other conditions as a sign you have EDS because they occur together so often.
Fibromyalgia
EDS can cause considerable chronic pain, especially hEDS, though because the other types of EDS are less, it’s hard to study how much chronic pain the other types can cause. Your EDS chronic pain is often misdiagnosed as fibromyalgia.2 The three major symptoms of fibromyalgia include widespread pain, fatigue and cognitive trouble like short-term memory issues. Notably, the pain you feel with fibromyalgia is primarily in your muscles and other soft tissue. While you can have pain anywhere with EDS, it’s more likely to center around your joints. There isn’t an accurate test for fibromyalgia yet, but EDS can be diagnosed via genetics or by closely following the comprehensive hEDS diagnostic criteria outline.
Related:
Mast Cell Activation Disorders
Your doctor may want to check for mast cell activation disorders.12, 14 Mast cells are a type of blood cell that release many immune-regulating mediators like histamine, chromogranin A, heparin and tryptase. If you have a mast cell activation disorder, these cells go haywire and respond incorrectly to internal triggers, leading to allergy symptoms such as itching and rashes that impact a number of systems in your body.
Mast cell activation is a more controversial and developing area of research. Based on the latest available information, it is more likely you will experience histamine-related allergies and intolerances as opposed to mast cell activation or mast cell activation disorder.
Related:
Chronic Fatigue Syndrome
Chronic fatigue syndrome typically has three major symptoms that may sound familiar — fatigue, chronic pain and trouble thinking clearly. Chronic fatigue syndrome is another condition your doctor may consider along with EDS, but chronic fatigue is a common EDS symptom. This is because while you can strengthen your muscles, you can’t strengthen your ligaments that hold bones together or tendons that attach muscles to bones. If you have hEDS and cEDS especially, you likely will have extra muscle fatigue because your muscles have to do three full-time jobs (as muscles, tendons and ligaments) 24/7 that can lead to chronic fatigue.
Learn More About Ehlers-Danlos Syndrome: Overview | Symptoms | Treatment | Resources

Sources
- Atwal, P. (2018). Ehlers-Danlos Syndrome [Telephone interview].
- Blitshteyn, S. (2018). Ehlers-Danlos Syndrome [Telephone interview].
- Brady, A. F., Demirdas, S., Fournel-Gigleux, S., Ghali, N., Giunta, C., Kapferer-Seebacher, I., … Malfait, F. (2017). The Ehlers-Danlos syndromes, rare types. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 175C, 70-115. Retrieved from https://doi.org/10.1002/ajmg.c.31550
- Chaplin, A. (2018). Ehlers-Danlos Syndrome [Telephone interview].
- Ehlers-Danlos Society. (2017). EDS Diagnostics 2017. Retrieved from https://www.ehlers-danlos.com/eds-diagnostics/
- Ehlers-Danlos Society. (n.d.). Assessing Joint Hypermobility. Retrieved from https://www.ehlers-danlos.com/assessing-joint-hypermobility/
- Ehlers-Danlos Society. (2017). Hypermobile Ehlers-Danlos syndrome (hEDS) vs. Hypermobility Spectrum Disorders (HSD): What’s the Difference? Retrieved from https://ehlers-danlos.com/wp-content/uploads/hEDSvHSD.pdf
- Genetic and Rare Diseases Information Center. (2018). Stickler syndrome. Retrieved from https://rarediseases.info.nih.gov/diseases/10782/stickler-syndrome
- Genetics Home Reference. (2018). How is genetic testing done? Retrieved from https://ghr.nlm.nih.gov/primer/testing/procedure
- Malfait, F., Francomano, C., Byers, P., Belmont, J., Berglund, B., Black, J., . . . Tinkle, B. 2017. The 2017 international classification of the Ehlers–Danlos syndromes. American Journal of Medical Genetics Part C, 175C: 8–26. Retrieved from https://doi.org/10.1002/ajmg.c.31552
- Marfan Foundation. (2018, October 03). What is Marfan Syndrome? Retrieved from https://www.marfan.org/about/marfan
- Mast Cell Action. (n.d.). About MCAS. Retrieved from https://www.mastcellaction.org/about-mcas
- Tinkle, B., Castori, M., Berglund, B., Cohen, H., Grahame, R., Kazkaz, H., & Levy, H. (2017). Hypermobile Ehlers-Danlos syndrome (a.k.a. Ehlers-Danlos syndrome Type III and Ehlers-Danlos syndrome hypermobility type): Clinical description and natural history. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 175C, 48-69. Retrieved from https://doi.org/10.1002/ajmg.c.31538
- Riley, B. (2018). Ehlers-Danlos Syndrome [Telephone interview].
- What is the cost of genetic testing, and how long does it take to get the results? – Genetics Home Reference – NIH. (2018). Retrieved from https://ghr.nlm.nih.gov/primer/testing/costresults
The Mighty’s Condition Guides combine the expertise of both the medical and patient community to help you and your loved ones on your health journeys. The guides are living documents and will be updated with new information as it becomes available.
condition_guides