Darwin’s Orphan: A Childhood With Epidermolysis Bullosa

Editor's Note
In this three-part Mighty feature, we explore what it really means to have an “orphan disease” — the name for conditions that affect less than 200,000 people in the U.S. There are 7,000 orphan diseases, collectively affecting 10 percent of the U.S. population.
Part One followed John Hudson Dilgen, a teenager with epidermolysis bullosa (EB), an extraordinarily painful orphan disease. Part Two widened the lens to explore orphan diseases more broadly from the clinical and policy perspective. Part Three turns the camera around, to consider the similarities, today and tomorrow, between orphan and common diseases. It concludes: we are not so different.
Part One: Him
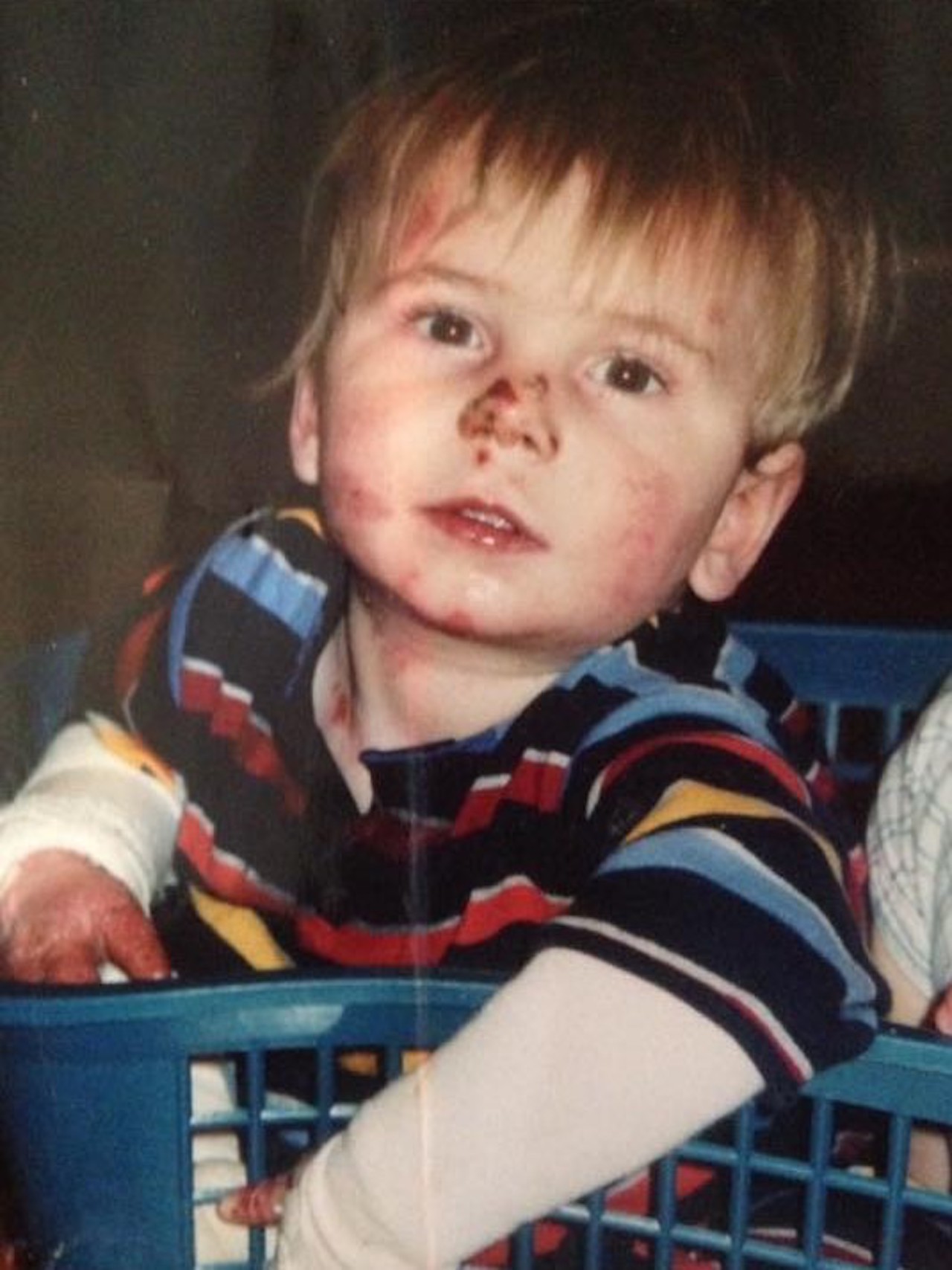
John Hudson Dilgen hates baths, but he does take them. Lots of them. Every other day, in fact. Yet, while most throw a toe to the surface imagining the possibility of chill or burn, he does so knowing the inevitability of pain. That’s because his baths contain bleach, and many surfaces of his body lack skin. Given the small ulcers on his arms, the intermediate ulcers on his legs and that huge ulcer on his back, none of which have healed over the course of years, nor have any plans to do so in the near future, bleach is the primary means of preventing the inexorable sequence of infection, sepsis and death. So, every other day, John submerges resentfully into the bathwater.
The cause of John’s ulcers is a so-called “orphan disease” named epidermolysis bullosa (EB). Known colloquially as “butterfly boys,” children with this condition have skin that shears under the slightest pressure, not unlike a butterfly shedding its cocoon before taking flight. Elegant, if not devastating, transformation is an appealing metaphor until realizing John’s trajectory is degenerative rather than ascendant.
This experience is not John’s alone. It is not exclusive to EB. Orphan diseases in the U.S. are defined as those affecting fewer than 200,000 individuals. There are up to 7,000 orphan diseases, and, collectively, they affect up to 10 percent of the U.S. population, or some 35 million Americans (1). This is six times the U.S. occurrence of Alzheimer’s disease, more than double the number of American cancer patients and involving 5 million more Americans than does Type-2 diabetes (2-5).
Globally, 350 million individuals have an orphan disease, roughly the size of the entire U.S. population (1). This is more than 10 times the number of people in the world with HIV and involves some 130 million more per year than malaria does (6-8). The literature refers to orphan diseases interchangeably as rare diseases. That, statistically, seems inaccurate.
John is both unique and very much not so. His scars are a microcosm of the pain sustained by patients facing analogous circumstances elsewhere, cornered by insurmountable hurdles at the scientific and systemic levels alike. A thorough probing of his ulcers, their ulcers, our ulcers, may be both diagnostic and therapeutic at once.
***
I met John on the third hour of the second day of my first week in medical school at New York University, when he was wheeled into Alumni Hall auditorium. We were learning about cell biology — specifically, the links that anchor skin to underlying tissue — and he was our “case model.” Put differently, he was a personification of pathology. Put differently, he was a patient. These are all synonyms, really.
John was my first patient. In the time that followed, he thoroughly rebuked my flawed expectations: maturity, hope and determination superseded misery, hopelessness and desperation. I wanted to help him.
But it wasn’t until three months later that I called his mother, Faye. Soon thereafter I found myself on a journey to the tail of Staten Island, seeking out an orphan disease in the orphaned borough.
***
Out the window of the subway car, rickety houses line the streets. A stray dog lopes across the road, vines veil boarded-up factories, and dented, rusty trucks bundle together in lots. The mottled, organic imperfections of the human condition express themselves through the architecture here. Indeed, Staten Island is in pain, physically and existentially: a storm of opioid addiction is sweeping across the island, killing hundreds and hospitalizing thousands over the past several years (9). It seems an appropriate place to seek the medically outcast.
Tottenville, John’s community, has ranked in the top five of New York City neighborhoods with the highest rate of opioid deaths for two years running (10). I get off at the last stop (“The End,” a sign reads) and walk down quiet side streets to the Dilgens’ colonial-style bungalow. Faye beckons me in. John reclines on a heavily padded, dilapidated armchair in front of the TV. “The Price is Right” is on. A throw blanket with NHL team logos hangs off his back. John, 14 years old and two months a middle school graduate, was recently named amongst the 50 Most Influential residents of Staten Island. I am a guest in the royal court.
He reigns, the bandages running down his arms and legs poorly concealed. Only his hands and head remain uncovered. It is a strange kind of regalia, for an unusual kind of prince, presiding over a unique kind of fame. As with most princes, this throne was his birthright, this kingdom his mandate. EB is genetic.
He is pale and slim. His chestnut brown eyes effuse wisdom beyond their years. His father, John Sr., sits on the couch next to us. His golden retriever, Dash, and his spotted kitten, Romeo, lie at his feet. The court’s jester, Bob Barker (it’s a rerun), is underwhelming across from him. John seems used to the rabble.
***
One way of conceptualizing John’s condition is through his appearance. John sketched his self-portrait:
My body is covered in bandages, and everywhere there is a bandage, there’s a wound. My feet have wounds, my shins, my thighs. My arms have wounds. My back, that would be the worst; my whole back is like a sheet of wounds.
Alternatively, consider his EB by the structure of his day. He starts his morning by taking innumerable medications. The current tally is somewhere between 15 to 18, John Sr. clarifies. Each year, he receives a couple more: some for his anemia, some for his depression, some for his analgesia and some for his analgesia-induced headaches (on the headaches: “It’s funny, the only thing that even helped were the mints.”). Dad also clarifies, grinning, that only one kind of mint, Icebreaker Duos, works. The Dilgens have a bulk purchasing deal at the local candy store.
The agenda of a day depends on when the bath is because John is usually exhausted thereafter. When the bath is, in turn, “depends on when [the nurses] come. Sometimes I’ll have a bath at 9:30 in the morning, which pretty much ruins the afternoon.” After the bath, he rests “for a couple hours, so I can do things later,” like eat dinner downstairs.
Otherwise, consider John’s condition through his experience of pain. His pain is constant, though alternatively dull and sharp, throbbing and stabbing. It is unpredictable. Sometimes the small wounds, or the large ones, will hurt; sometimes the new or the old; sometimes those that excoriate the fastest, or the slowest. Previously, he had used a cup to pour water during his baths because the faucet pressure was too high. He elaborated, “Recently, we got a new shower head that sprays mist, but even the mist, it’s not like mist mist.”
He takes his current painkiller, Dilaudid, as often as he can. Pharmacologically speaking, every four hours. He settled on Dilaudid after trying 10 other analgesics because he could feel it. It takes 45 minutes to kick in, providing about two hours of relief (Icebreaker Duos are administered). Relief diminishes his pain from a nine to a seven out of 10. His experience of pain is inverted from that of normal: analgesia being the exception, rather than the norm. He nodded in affirmation of the calamity. Faye and John Sr. shook their heads. Dash and Romeo yawned. Barker chortled.
Finally, attempt to fathom John’s EB through his relationship with school. Or, his lack thereof. John missed both sixth and seventh grades because he could not sit through a day of class. More than a couple of hours in a school chair efficiently and unceremoniously tore the skin off his buttocks. He recounted: “I tried to start sixth grade. But sitting in class, my body was aching. I couldn’t focus. I tried. I just couldn’t. ”
In lieu of school, there are few things to occupy the vast number of hours. One gains a newfound appreciation for the virtue of school when these basic structures are subverted. Even knowing John has a 60-percent chance to walk, alive, at graduation.
As such, he spends ample time with Romeo, “a nice distraction,” and Dash, “a great companion.” Distraction provides welcome variety from the placidity of elapsing time. The one command the Dilgens taught Dash, outside the trainer’s jurisdiction, was “get Mom.”
***
Faye Purpura Dilgen is endlessly energetic and unapologetically enthusiastic, with the paired patience of a psychiatrist and impatience of an innovator. This serves her well in her working life as a physical therapist and in her family life as a caregiver.
The title of caregiver, for a mother of a child with an orphan disease, is universal: giving care, in the absence of a cure, involves every step from acquisition to administration. The vocabulary of the caregiver, ranging from allograft to zymogen, is equally vast. Thirty-eight percent of parents of children with orphan diseases must reduce their workload to provide care, and one-third cease working entirely (11).
Faye grew up in a religious Catholic family: one brother is a priest, and one aunt is a nun. Sitting with her feels like sitting with Mother Mary. On the question of her relationship with God, as the mother of a child with an incurable disease, she declines “that it changed anything.” Rather, her interaction with religion is more pragmatic. She prays, “Firstly for a cure. In the meantime, for comfort and if something happens, that it happens quickly and painlessly.”
She recounts the story of receiving John’s diagnosis. In the seconds following birth, those huddled around the newborn noticed a region of skin absent at his left heel. In the minutes thereafter, the nurses placed EKG leads on his back, which subsequently tore the skin off there, too. In the hours that followed, the clinicians grew suspicious and workup for EB began. In the coming days, biopsies were taken and luminescent stains came back positive. A week after diagnosis, she received her first, “Congratulations on the baby boy,” from an EB specialized case-worker.
In the same way, she describes her effort to alter John’s prognosis. The caregiver’s role to this end is subclinical: responsible for the regimented, mundane, non-academic activities of day-to-day maintenance outside of the specialized healthcare setting. That is, ensuring John’s survival with each passing 24-hour interval. The hope is for optimization of health along the existing trajectory, until therapies emerge capable of rerouting. It is a war waged against natural history of disease, using the firepower available through standard of care.
The campaign started immediately following diagnosis: Faye was attempting to use a dressing as instructed on the label, but John’s pain was excruciating. She appealed to one of the few EB support groups at the time. Someone asked, “What are you buttering it with?” She answered that she doesn’t butter, it doesn’t say to butter, and, oh yeah, what is buttering? They wrote back again: “Use 1/3 coconut oil, 1/3 Aquaphor, 1/3 Medihoney, mix it, slather it, fold it in half, and put the primary layer underneath.” The notion that “it takes a village to raise a child” is especially true for children with orphan diseases. The recipes shared by mothers in the village extend beyond the kitchen.
Simultaneously, Faye does her darnedest to make John comfortable and make him happy. Even dinner, the stalwart of the calendar, is a complicated chore, since EB affects the lining of John’s GI tract, and John’s analgesics give him stomach aches. In Faye’s words, he faces “an eternal conflict between being hungry and not wanting to eat.” Mac n’ cheese is a staple, and in situations where John develops an esophageal stricture (due to scarring), it serves a diagnostic role: if he can’t swallow it, he flies as an emergent case to the University of Cincinnati for dilatation. In celebratory circumstances, John’s favorite food is crab legs, which his mother dutifully deconstructs bedside following a successful surgery.
Nonetheless, immaculate though she may be, Faye’s ability to alter the trajectory of John’s life is limited.
The ability to alter the sequence of events at the bedside is, scientifically, derived from the bench. To combat, much less conquer, disease, a tactician must first understand its nature.
The nature of epidermolysis bullosa summons from the dysfunction of a protein called Collagen VII, a microscopic bridge yoking cells together. At the skin (epidermis), it anchors the surface layer to the basement membrane (dermis), providing resilience amidst trauma. Without Collagen VII, the epidermis yields (lysis), the dermis is exposed, and blisters arise (bullae) (12-15).
One of the champions of the bench is Jakub Tolar. Originally from the Czech Republic, Dr. Tolar now works at the University of Minnesota. Formally trained as a hematologist and a pioneer of gene therapy, Dr. Tolar began his work on EB after a mother who “broke in cradling her son” confronted him at a conference. She knew his work and insisted he “has to figure out how to make this work,” pushing her son into his spaghetti-limp arms.
Dr. Tolar dons an unbranded pair of narrow black glasses that frame his perspective on the role of a physician, a role fundamentally requiring “humility, in the face of things you’ve been called upon to combat.” His combat objective is immodest and uncompromising: to “open a future that has not existed before.” Appropriately, his research, which re-engineers the genetic code of epidermal cells to produce Collagen VII, could do just that for patients with EB.
John could benefit greatly from a future, not least because he has a 40-percent chance of dying before the age of 20. He will most likely die from metastatic skin cancer (13). A scientist is naught but for the ripples he creates, and Dr. Tolar is aiming for a tsunami.
Ripples, though, are unpredictable. ”Great science,” Dr. Tolar laments, “is oblique.” Serendipity and discovery are products of circumstance rather than planning. Thus, rather than camping out behind a bush waiting for the declaration of gravity, a scientist must play Newton, placing themselves in a position sufficiently vulnerable as to let the apple fall. Scientific breakthroughs necessitate a resignation to the chaos; the apple of knowledge is consumed by way of observation rather than insistence.
Dr. Tolar acknowledges the depraved irony. Nature is incomprehensible in its complexity, yet experimental elimination of confounding variables is, by design, an exercise in fabrication. Alas, the academic research industry of grants and tenure depends on narrow hypotheses, artificial environments and precise objectives. It has little tolerance for failure.
This dynamic has contributed to the well documented “publication bias” against negative results (16). Results in the negative are often disregarded as failed hypotheses rather than successful observations — and subsequently, not accepted for publication. Illustrative: in trials for antidepressants, 94 percent of those published were positive, whereas 50 percent of those assessed by the FDA were positive (17).
The so-called “reproducibility crisis” similarly derives from twisted incentives favoring productivity over transparency (18). The result is that, as John Ioannidis wrote in 2005, “most published research findings are false,” and 85 percent of effort therein is wasted (19, 20). In 2016 he concluded that “most clinical research is not useful”(21). Up to three-quarters of patients with a given disease do not respond to drugs indicated for them (22).
“We can’t ask [EB] patients to have patience,” exhorts Dr. Tolar, “they are not privileged with time.” Yet, he studies a disregarded disease in a derelict pocket of the Northwest subject to a system ill-suited for facilitating breakthrough science. Tip-toe incrementalism is incentivized over innovation. Urgency is lacking. John’s clock ticks. At least there are Icebreaker Duos?
***
Read Part Two and Part Three of this series.
Photo shared with permission of Faye Dilgen.